
Elizabeth M. McNally and Peter Pytel
Department of Medicine, Section of Cardiology, University of Chicago, Chicago,
Illinois 60637; email: emcnally@medicine.bsd.uchicago.edu
Department of Pathology, University of Chicago, Chicago, Illinois 60637;
email: Peter.Pytel@uchospitals.edu
Link of original article:
Key Words
myotonia, sarcopenia, muscle regeneration, dystrophin, lamin A/C,
nucleotide repeat expansion
Abstract
Dystrophic muscle disease can occur at any age. Early- or childhoodonset muscular dystrophies may be associated with profound loss of muscle function, affecting ambulation, posture, and cardiac and respiratory function. Late-onset muscular dystrophies or myopathies
may be mild and associated with slight weakness and an inability to increase muscle mass. The phenotype of muscular dystrophy is an endpoint that arises from a diverse set of genetic pathways. Genes associated with muscular dystrophies encode proteins of the plasma
membrane and extracellular matrix, and the sarcomere and Z band, as well as nuclear membrane components. Because muscle has such distinctive structural and regenerative properties, many of the genes implicated in these disorders target pathways unique to muscle or
more highly expressed in muscle. This chapter reviews the basic structural properties of muscle and genetic mechanisms that lead to myopathy and muscular dystrophies that affect all age groups.
MUSCLE STRUCTURE
The basic cellular unit of voluntary muscle is the myofiber. Myofibers are elongated,
multinucleate cells. During development and regeneration, myofibers arise from the fusion
of singly nucleated myoblasts to form myofibers. Individual myofibers are encased in an
endomysial sheath of connective tissue, and groups of myofibers are encased in epimysial
connective tissue (Figure 1). The ends of myofibers form myotendinous junctions, a
specialized attachment for bony insertion that can withstand considerable force. The cytoplasm
of each individual myofiber is highly organized, containing chains of sarcomeres
that run parallel to the length of the myofiber. Electron-dense Z bands define the borders
of each sarcomere, and individual sarcomeres are composed of actin-containing thin filaments
and myosin-containing thick filaments (Figure 2). The heavy chain of myosin hydrolyzes
ATP to provide the energy for muscle contraction. The rate at which myosin hydrolyzes
ATP is proportional to the speed of muscle contraction. Slow type I and fast type
II fibers express different myosin isoforms and can be distinguished by enzyme histochemistry
in the ATPase reaction (Figure 1d ).
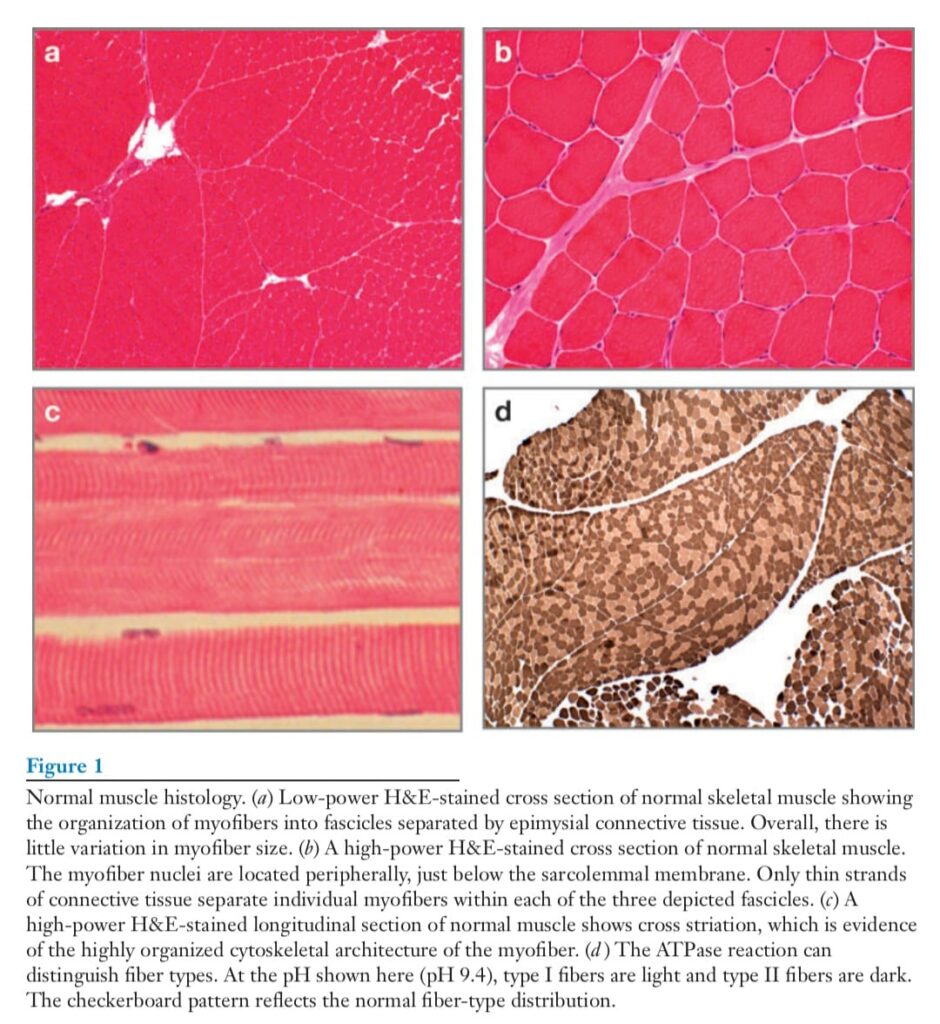
The fiber type is determined by the innervating motor neuron in the spinal cord. The motor unit is the functional unit of one motor neuron and all the myofibers innervated by it. Several groups of muscle diseases present as weakness, cramping, or muscle pain. These include the congenital myopathies, the muscular dystrophies, myotonic disorders, storage diseases, mitochondrial diseases, and inflammatory myopathies. These primary muscle diseases are distinct from neuropathic diseases and disorders of neuromuscular transmission. This review focuses on myopathies arising from defects of cytoskeletal, sarcomeric, and membrane-associated proteins of the plasma membrane and the nucleus (Table 1). Historically, the diseases that fall into this group were classified on the basis of features such as the pattern of inheritance, the age of disease onset, and the primarily affected muscle groups. The increasing ability to link these entities to specific genetic defects has greatly increased our understanding of these diseases.

MUSCLE DEGENERATION AND REGENERATION
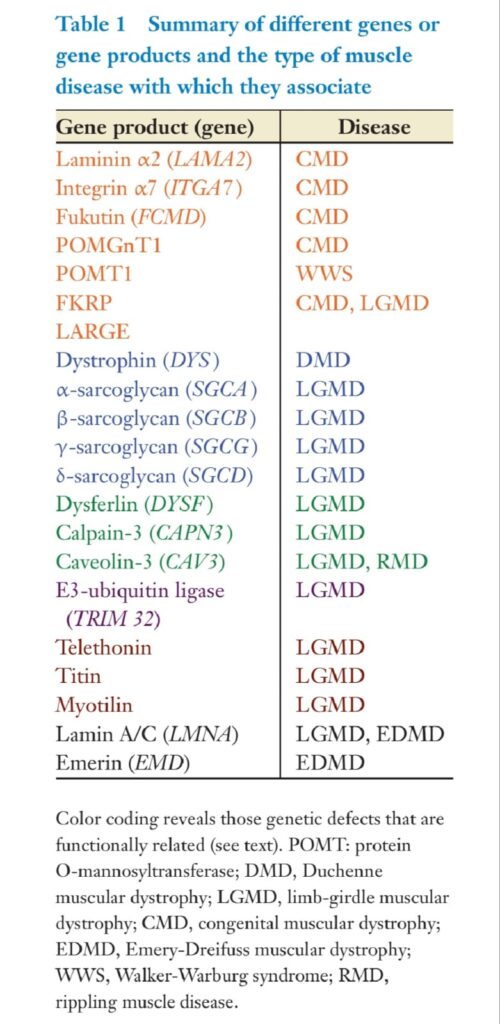
Muscle damage typically takes the form of myofiber necrosis and regeneration (Figure 3).
This process can be segmental, involving only a part of an individual myofiber. The necrosis is associated with membrane damage and leakage of cytoplasmic proteins, such as creatine kinase (CK) and lactate dehydrogenase, that can serve as serum markers of muscle damage.
The basement membrane of the necrotic fiber remains as a scaffold for the regenerative
process. Muscle is highly regenerative. The regenerative process occurs with the organization
of necrotic cytoplasmic debris by inflammatory cells. Depending on the degree of damage, the time course of muscle regeneration occurs over one to three weeks. Mature muscle contains mononuclear cells that reside between the basement membrane and the plasma membrane or sarcolemma of each myofiber. The nuclei of these mononuclear cells selectively take up bromodeoxyuridine, indicating mitotic activity. These two features, the mitotic activity and the location between the plasma membrane and the basal lamina, were used to define muscle satellite cells (1). Experimental evidence supports that the origin of the satellite cell is the myotome (2), but additional evidence suggests that bone marrow–derived cells may contribute to muscle regeneration (3). Satellite cells are activated by injury. As quiescent cells, satellite cells divide and maintain the satellite cell pool. With injury, satellite cells activate and then differentiate into myoblasts (for a review, see Reference 4). Myoblasts represent a committed cell that will eventually withdraw from cell cycle activity and express genes found in myotubes.
Myoblasts can fuse to each other or to existing myotubes to generate new muscle. The nuclei from myoblasts that have recently fused to an existing myofiber are found in the center of myotubes.Within one to three months, these nuclei will assume a peripheral position near the sarcolemma, as is characteristic of the mature myofiber. Because most muscle disease involves
myofiber damage or degeneration, regeneration is a feature of all muscle disease. Although
central nucleation is seen in normal muscle fiber, an increase in the number of centrally
nucleated myofibers may indicate an ongoing degenerative or necrotic process. The muscle
biopsy, viewed perpendicular to the long axis of the myofiber, remains the gold standard for
evaluating muscle disease. The main muscle biopsy changes found in myopathic diseases
are the presence of necrotic myofibers and regenerative myofibers. Necrotic myofibers
contain variable amounts of amorphous cellular debris, inflammatory cells, and satellite
cells. Regenerative myofibers are characterized by their enlarged nucleus and basophilic
RNA-rich cytoplasm. After the reconstitution of the sarcomeric myofiber architecture, the
myofiber nuclei maintain a centralized position away from their normal subsarcolemmal
location for several weeks. In addition, myopathic diseases also show an increased variation
in myofiber size with regenerating fibers that tend to be smaller, and with unaffected
fibers that may show compensatory hypertrophy. In diseases associated with prolonged,
continued myofiber injury, the regenerative process fails to maintain normal skeletal muscle
architecture. In these cases, increased connective tissue in the form of interstitial fibrosis
and fatty replacement are evidence of the chronicity of the disease (Figure 3).
CONGENITAL MUSCULAR DYSTROPHIES
The congenital muscular dystrophies (CMDs) are apparent at birth, manifesting frequently as a “floppy” infant lacking muscle tone. CMD is a feature of Walker-Warburg Syndrome, muscle-eye-brain disease, and Fukuyama-type CMD. In these disorders, additional neurologic features such as lissencephaly and ocular and retinal defects may occur and may reflect a failure of
neuronal migration. The genes mutated in the CMDs encode enzymes that contribute to O-linked glycosylation (5–9). In general, asparagine-linked glycosylation is common to more proteins, with only a minority of proteins undergoing O-linked glycosylation—usually on serine residues. A major target protein affected by these diverse genetic disorders is α-dystroglycan, as it is known to undergo O-linked glycosylation (10, 11). As discussed in more detail below,
dystroglycan is a core component of the dystrophin glycoprotein complex (DGC) (12).
Dystroglycan is composed of two subunits produced from a single gene (Figure 4).
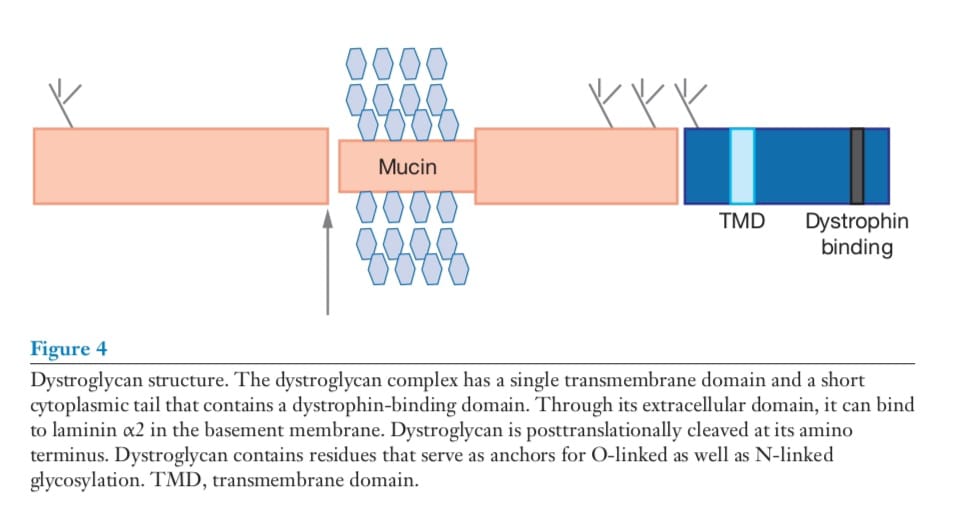
Dystroglycan is broadly expressed, and α-dystroglycan is variably glycosylated with tissue specificity (13). β-dystroglycan is a transmembrane protein that anchors α-dystroglycan.
Although extramuscular involvement is common in CMDs, not all genes in this class lead to extramuscular involvement. For example, mutations in the gene encoding the fukutin-related protein (FKRP) associate more with muscle defects with less severe or absent central nervous system findings (14, 15). The degree to which there are central and peripheral nervous system findings can be partially explained by the specific responsible mutations.
As such, genotype-phenotype correlation in these disorders may, to some degree, be predicted by the amount of glycosylated α- dystroglycan present. In CMDs, muscle has a dystrophic appearance and, consistent with this, patients may have an elevated serum CK, indicating disruption of the sarcolemma (16). Immunostaining of muscle biopsies with antibodies directed at core residues in α- dystroglycan may appear normal, as may antibodies directed at β-dystroglycan. Immunostaining with antibodies directed at the glycosylated forms of α-dystroglycan generally show a reduction proportional to the severity of disease. The decrease in α- dystroglycan glycosylation may be nonuniform along the length of the myofiber. The genes associated with CMDs are thought to encode glycosyltransferases (Figure 5) (17, 18).
Where antibodies have been generated, it appears these proteins are resident in the Golgi apparatus or in the endoplasmic or sarcoplasmic reticulum (19–22). One form of CMD arises from mutations in the gene encoding the α2 chain of laminin (23, 24). In muscle, the major basement protein laminin is composed of the α1, α2, and γ chains to form merosin or laminin-2. Although the defect here is not one that affects glycosylation of dystroglycan directly, merosin is a major ligand for α-dystroglycan; thus, disruption between the extracellular matrix and the membrane occurs, albeit by a mechanism distinct from that in the CMDs listed above.
THE MUSCULAR DYSTROPHIES
Duchenne Muscular Dystrophy
Muscular dystrophies in children or adults are characterized by a progressive muscle weakness that may have a predilection for certain muscle groups. Childhood-onset muscular dystrophies are usually lethal, owing to associated cardiac muscle or respiratory muscle weakness. Duchenne muscular dystrophy (DMD) represents the most common Xlinked inherited disorder. DMD is produced by mutations in the dystrophin gene on the X chromosome, and most affected boys are diagnosed in the first few years of life. DMD boys display delayed walking, falling, a toe gait, and calf hypertrophy. Commonly, serum CK levels are substantially elevated. Gross deletions or duplications in the dystrophin gene account for 60% of mutations, and these mutations are detected by a PCR-based blood test (25). The efficacy of this test is such that many DMD boys are no longer subjected to muscle biopsy.
As much as 40% of DMD patients do not have a large gene deletion or duplication; instead, a point mutation is responsible for their disorder (26). A number of these point mutations may insert novel stop codons, and this is the mechanism for the dystrophic phenotype in the mdx mouse model. The mdx mouse displays many of the histopathologic features seen in DMD, but these mice—unlike their human counterparts—remain ambulatory and have only a mildly reduced lifespan. A point mutation in exon 23 creates a novel stop codon and truncation of the dystrophin protein (27). Recent strategies for treatment design target stop codon read-through as a mechanism for treating these DMD patients (28). At least 10% of DMD patients may be
treated with this approach. Novel agents with improved read-through capabilities and a reduced side-effect profile are being developed and tested. These compounds are associated
with a systemic delivery that leads to an increase of 10–30% over normal dystrophin protein levels (29). Gene deletions that only partially disrupt dystrophin protein expression are usually associated with the milder phenotype of Becker muscular dystrophy (BMD). Many of these
mutations produce internally deleted dystrophin that lacks spectrin repeats but maintains the core actin-binding and carboxyterminal regions. For many BMD patients, a muscle biopsy may be helpful in providing a diagnosis that may be essential for genetic counseling. Maternal carriers of DMD-associated dystrophin gene mutations can show symptoms of mild muscle weakness. Cardiomyopathy may manifest in female dystrophin mutation carriers owing to
X-inactivation, which affects the wild-type dystrophin locus that remains in these women
(30).
Dystrophin is a large protein and a central component of the DGC that provides stability to the sarcolemma (Figure 6).
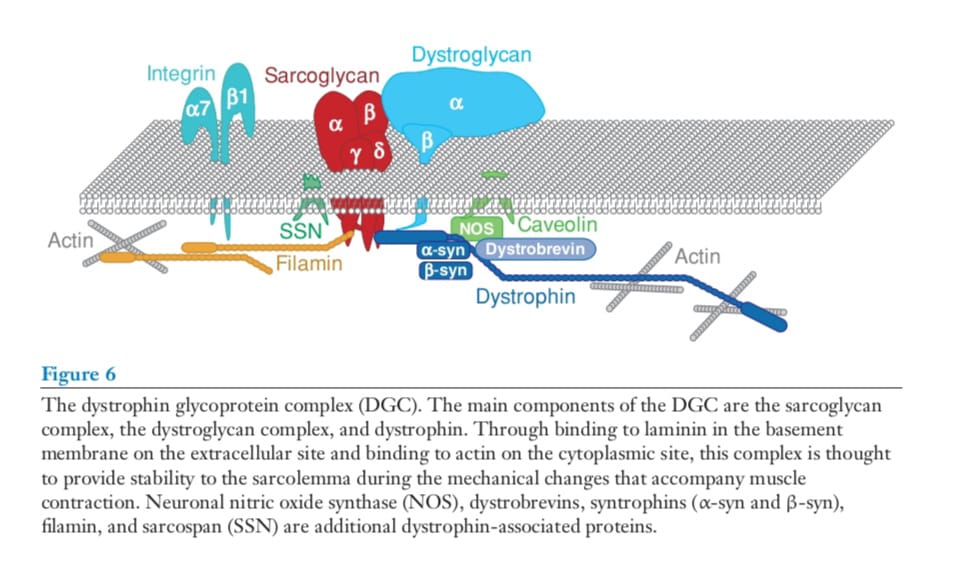
Dystrophin has a calponin-like actin-binding domain at its amino terminus and 24 spectrin repeats interrupted by four hinge points (31). The carboxyl
terminus of dystrophin binds β-dystroglycan directly (32). The amino terminus and regions
along the spectrin-repeat rod domain bind to cytoplasmic γ-actin, forming a mechanically
strong link (33). In the absence of dystrophin, muscle contraction enhances membrane disruption and produces myofiber damage. Disruption of the myofiber can be imaged by
uptake of the vital tracer Evans blue dye (34, 35). Gene replacement experiments have
identified a minimal region of dystrophin required to protect muscle against contractioninduced damage (36). These experiments indicate that an intact actin-binding domain, four spectrin repeats, and the carboxyl terminus are needed to replace dystrophin function. Membrane disruption that arises from the loss of dystrophin leads to an increase in intracellular calcium content, which promotes a series of pathogenic events including calcium-activated proteolysis and sarcomere dysfunction (37). The carboxyl terminus of dystrophin links it to the remainder of the membraneassociated DGC (38, 39). The DGC includes cytoplasmic and transmembrane components, and the entire complex encompasses mechanical and signaling roles (40, 41). The cytoplasmic components of this complex include the dystrobrevins, small proteins that bind
directly to dystrophin, and have homology to the carboxyl terminus of dystrophin. The
PDZ domain-containing syntrophins bind to dystrobrevin and to neuronal nitric oxide synthase (also known as nNOS or NOS1). In the absence of dystrophin, the cytoplasmic DGC components, in addition to the transmembrane components dystroglycan and glycan, are destabilized at the sarcolemma. The loss of dystrophin leads to the displacement of nNOS from the sarcolemma, and this displacement mediates abnormal contractioninduced vasorelaxation (42, 43). The relationship of this loss to disease pathogenesis is not entirely clear because the loss of nNOS from the plasma membrane is insufficient to produce a dystrophic phenotype (44). It is more
likely that the loss of nNOS from the membrane plays a contributory role in that it may augment tissue damage in response to dystrophin loss. In DMD, the muscle biopsies show myopathic changes with myofiber degeneration and regeneration (Figure 7).
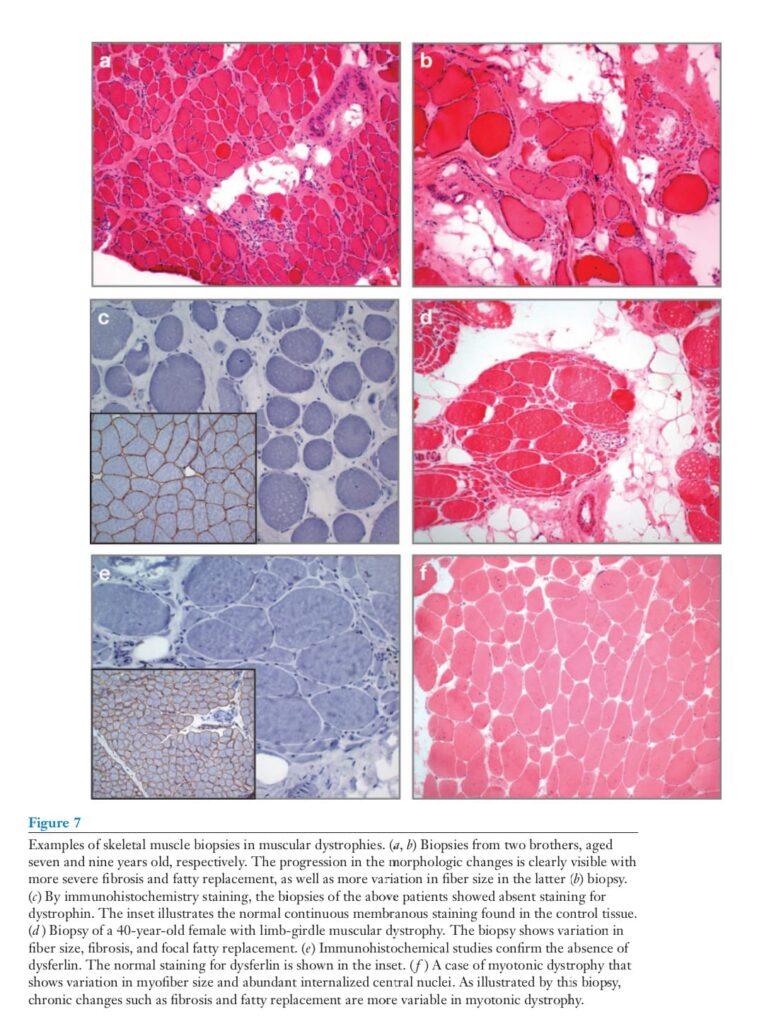
With disease progression, the muscle shows increasing fatty replacement and endomysial fibrosis. Immunohistochemistry demonstrates the absence of the normal sarcolemmal staining for dystrophin. Immunostaining biopsies of DMD patients for the remaining DGC components reveal secondary deficiencies of the sarcoglycans, dystroglycan, and the syntrophins as evidence of the incomplete assembly of the remaining DGC in the absence of dystrophin. In BMD, the biopsy changes develop at a much more protracted pace, and immunohistochemical staining may only show deficient staining for certain domains of the protein.
Limb-Girdle Muscular Dystrophies:
The Sarcoglycans
The sarcoglycans are transmembrane elements that form a tight unit within the DGC
(40, 45). There are at least six sarcoglycan proteins, although the major sarcoglycan complex found at the muscle membrane is composed of four sarcoglycan proteins, α, β, γ, and δ. Mutations in these genes are the underlying defects in a subset of the recessively inherited limb-girdle muscular dystrophies (LGMDs). ε-sarcoglycan was identified on the basis of its high homology to α-sarcoglycan, and mutations in the gene encoding ε-sarcoglycan lead to myoclonic dystonia (46). ζ -sarcoglycan was identified on the basis of its homology to γand δ-sarcoglycan, and mutations in this gene have not been described (47). Patients with LGMD due to sarcoglycan gene mutations have a presentation similar to the phenotypic range seen in DMD and BMD. Mutations in the α-sarcoglycan gene are frequently point mutations that may associate with milder phenotypes (48). Frameshifting and select point mutations may lead to severe phenotypes. There is a common frameshifting deletion in the gene encoding γ-arcoglycan
that has been described in many distinct populations consistent with a common disease allele (49). The identical mutation in the γsarcoglycan gene can produce a phenotype of varying disease severity in humans and in mouse when placed in different genetic backgrounds. These findings suggest that genetic modifiers may influence the phenotypic outcome in the muscular dystrophies (50). The basic pathogenic features in muscle biopsies from sarcoglycan mutant patients are indistinguishable from those found in DMD or BMD muscle. Antibody staining to the
sarcoglycan subunits is often all depleted in response to mutations in any single sarcoglycan
gene (51). An exception to this is with tions in γ-sarcoglycan, where residual sarcoglycan staining can be present but the phenotype may still be severe. α-sarcoglycan gene
mutations may also display residual sarcoglycan expression at the plasma membrane.
Mutations in sarcoglycan subunits generally do not affect the distribution of dystrophin.
Thus, mutations in dystrophin lead to the loss of the sarcoglycan subunits from the plasma
membrane, but the reverse is not true as sarcoglycan gene mutations leave dystrophin intact. As the phenotypes are equally severe from dystrophin and sarcoglycan gene mutations, it is therefore the disruption of the sarcoglycan complex, as the common molecular feature, that is critical for the dystrophic process. Mutations in γ- and δ-sarcoglycan produce a similar phenotype in mouse models. Despite phenotypic similarities, these two mutations do not cause similar disruptions in the sarcoglycan complex, but result in muscle damage through different pathways. Loss of δ-sarcoglycan causes contraction-induced muscle damage similar to that seen in dystrophin mutations (52). The γ-sarcoglycan mutation is not associated with the same type of mechanical damage, implicating a separate mechanism for the myopathic changes. These results indicate that the sarcoglycan complex may serve important functions beyond maintaining mechanical strength as part of the DGC. Smooth muscle dysfunction may be
present in DMD patients. However, there are differences between the DGC in striated
and smooth muscle. Notably, the sarcoglycan complex in vascular smooth muscle is
composed of ε-, β-, δ-, and ζ -sarcoglycans (53). Interestingly, vascular spasm can occur
in response to sarcoglycan gene mutations (54). Vascular spasm is thought to be most
pathogenic to the heart, affecting the coronary artery vasculature. Restoration of cardiomyocyte δ-sarcoglycan expression in the background of δ-sarcoglycan null animals was sufficient to eliminate coronary artery vascular spasm. Therefore, vascular smooth muscle defects in DGC mutations can arise from vascular smooth muscle cell extrinsic
processes (55).
Limb-Girdle Muscular Dystrophies:
Dysferlin
Dysferlin is the protein product of the LGMD type 2B locus and is a membrane-associated
protein with a long cytoplasmic domain (56). Dysferlin is not associated with the DGC
and plays a distinct pathogenic role when disrupted. Dysferlin is homologous to the
Caenorhabditis elegans protein fer-1, named for its role in fertilization defects in C. elegans
mutants (57). In fer-1 mutants, there is a defect in vesicle fusion to the plasma membrane of the maturing sperm, leading to fertilization defects (58). Specifically, in fer-1 mutants, there is an accumulation of submembranous vesicles. Similarly, in dysferlin mutant muscle, there is also an accumulation of submembranous vesicles (59–61). In a manner analogous to fer-1, dysferlin mediates membrane-resealing events required for the muscle membrane repair in mature muscle (62). Laser-mediated sarcolemmal disruptions are repaired much more slowly in dysferlin mutant muscle compared with normal muscle (61). In these studies, it was confirmed that sarcolemmal resealing in response to damage is a calcium-sensitive event. Interestingly, resealing in the absence of calcium occurred at the same slow pace as resealing in the absence of dysferlin. These findings are consistent with a role for dysferlin as the calcium sensor for vesicle fusion that mediates sarcolemmal resealing (63). The cytoplasmic domain of dysferlin contains six C2 domains, and C2 domains are implicated in calcium and phospholipid binding. The C2 domains of the synaptotagmins display homology to those found in dysferlin,
and synaptotagmins mediate the calcium sensitivity of membrane fusion events associated
with the neurotransmitter release at nerve terminals (64). The first C2 domain of dysferlin binds a mixture of phosphotidylserine and phosphotidylcholine only in the presence of physiologically relevant calcium concentrations (65). Calcium-dependent phospholipid binding is abolished by a point mutation in the first C2 domain of dysferlin, and this mutation produces muscular dystrophy. The closely related protein myoferlin is highly expressed in myoblasts undergoing fusion to myotubes, and its first C2 domain displays similar phospholipid-binding capacity to that seen for dysferlin (66). Mice lacking myoferlin display reduced muscle size, and in culture
myoferlin null myoblasts fuse less well and do not form large myotubes (67). These findings
implicate myoferlin in the membrane fusion events associated with myoblast fusion to existing myotubes. LGMD 2B muscle biopsies display findings similar to those associated with other
forms of muscular dystrophy, with the exception that an inflammatory infiltrate may be seen as a prominent feature with dysferlin gene mutations (68, 69). This finding may be so great as to mimic inflammatory myopathies such as what is seen in polymyositis or inclusion body myositis (70). A subset of biopsies may show a decrease in dysferlin staining, yet have a normal dysferlin gene indicating an alternative mechanism for reducing dysferlin and for producing muscle pathology. Miyoshi myopathy is a mild form of muscular dystrophy associated with dysferlin
gene mutations that selectively affects the gastrocnemius muscle but spares other musculature. The identical mutation can be associated with the more severe LGMD or Miyoshi myopathy, indicating that additional genes and/or environmental factors may contribute strongly to modulate the dystrophic process (71).
Limb-Girdle Muscular Dystrophies:
Calpain, Titin, Caveolin, TRIM32, Myotilin, and Telethonin
LGMD 2A is a recessive form of muscular dystrophy associated with homozygous mutation in the gene encoding calpain-3 (also known as p94). These mutations are common and lead to a progressive loss of muscle function (72). Mice lacking calpain-3 have been generated and recapitulate aspects of the human phenotype of LGMD. Calpain-3 likely has a number of proteolytic targets, but filamin C, a muscle-specific filamin, is cleaved by calpain-3 in a manner that alters the binding of filamin C to subunits of the sarcoglycan complex (73). Calpain-3 is important for sarcomere turnover, and its pathways are distinct and upstream from ubiquitin. In addition, calpain-3 binds directly to titin, the giant protein that spans sarcomeres (74). Mutations
in titin lead to LGMD and the milder tibial myopathy. The murine model muscular dystrophy with myositis (mdm) is associated with a mutation in titin’s NB2 domain that interacts with calpain-3 (75). Several other gene products have been implicated in the formation of muscular dystrophy, including caveolin-3, TRIM32, and myotilin. Caveolae are membrane invaginations that participate in localizing components and proteins in the membrane. Caveolin-3 is found inserted in the membrane, and dominant and recessive mutations are associated with muscular dystrophy, as well as with more mild disorders (76). Caveolin-3 may be selectively reduced in response to dysferlin gene mutations (77). TRIM32 is a ubiquitin ligase that binds to myosin and ubiquitinates actin and thereby participates in sarcomere recycling (78). Myotilin is a Z band–associated protein that binds to other Z band proteins, and defects in myotilin lead to dominantly inherited muscular dystrophy (79). The full role of myotilin is not appreciated,
but its pathogenicity may be related to what is seen from mutations in other sarcomereassociated genes such as telethonin or, potentially, α-actinin (80). In addition, a subset of these disorders is associated with nemaline myopathy, where an accumulation of nemaline rods can be found in the cytoplasm as an indicator of a pathologic disease process
(81).
MUTATIONS OF NUCLEAR
MEMBRANE–ASSOCIATED GENES
Emery Dreifuss muscular dystrophy (EDMD) is an X-linked disorder associated with progressive muscle weakness and contractures. With EDMD, cardiac involvement such as atrioventricular heart block is frequent. Mutations in the gene encoding the nuclear membrane protein emerin produce EDMD (Figure 8).
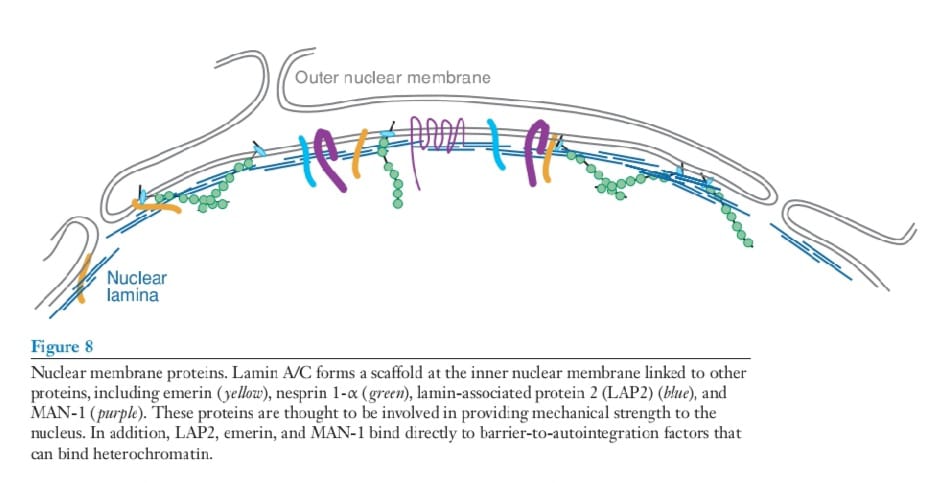
Emerin is a 34-kDa protein that embeds in the inner nuclear membrane.
Emerin contains within its primary structure an LEM domain, named for its presence in lamin-associated protein 2, emerin, and MAN-1. The LEM domain of emerin binds to
barrier-to-autointegration factor (BAF). BAF is small peptide that oligomerizes and directly
binds to DNA (82). Its position at the nuclear membrane coupled with its partnering
to BAF suggests that the inner nuclear membrane may play a role in scaffolding chromatin. Emerin is a broadly expressed protein, yet mutations in this gene primarily affect muscle (83).
Dominantly inherited forms of EDMD (AD-EDMD) are more common (84). Mutations in the gene encoding inner nuclear membrane protein lamin A/C (LMNA gene) are responsible for AD-EDMD. Lamins A and C are type V intermediate filament proteins that form much of the structural apparatus of the inner nuclear membrane of postmitotic cells (85). Lamins A and C dimerize and then form higher-order structures to provide tensile strength to the nucleus. In most postmitotic cells, lamins A and C dominate
the composition of the inner nuclear membrane intermediate filaments. In cells undergoing division, lamin B is more likely to be the dominant intermediate filament protein of the inner nuclear membrane. Exactly how mutations in broadly expressed genes lead to tissue-specific phenotypes that affect muscle is not known. Several nonexclusive hypotheses may explain this
phenomenon. LMNA gene mutations may render the nuclear membrane weakened such
that abnormal function develops when these nuclei are subjected to the force associated
with muscle contraction. This mechanical weakness hypothesis may explain some of the
susceptibility of striated muscle to mutations and defects in the nuclear membrane. Evidence supporting the mechanical hypothesis was found in murine cells engineered to lack
LMNA (86). In these homozygous null cells, maximal normalized nuclear strain was increased in LMNA null fibroblasts. Cytoskeletal stiffness was reduced and nuclear fragility was increased in LMNA fibroblasts. Interestingly, defective nuclear factor-kappa B (NFκB) signaling was associated with the loss of LMNA. These observations were made in fibroblasts from LMNA null mice, and therefore investigators argued that those cells most subject to stress and thus strain, such as muscle, would be the most adversely affected by loss of LMNA. A similar defect was associated with the loss of emerin (87). Many mutations in LMNA are not lossof-function mutations, but instead are dominant point mutations that may produce gainof-function activity and affect other attributes of the nuclear membrane. A second hypothesis to explain how LMNA gene defects target certain cells and tissues may relate to other nuclear functions perturbed by mutations in LMNA. The nuclear membrane regulates intracellular transport, DNA synthesis,
and gene transcription. The observation that many LMNA mutations preferentially target
postmitotic cells, such as myofibers and cardiomyocytes, may indicate defects in nuclear
transport or that gene expression may more likely explain the underlying cellular defect
associated with LMNA gene mutations. Not all LMNA mutations may lead to phenotype
through the same mechanisms. In support of this, recent data indicate that LMNA mutants
are associated with defects in skeletal muscle regeneration (88–90). The regenerative defect may explain aspects of the muscle phenotype seen in autosomal-dominant EDMD patients, but may not fully explain the accompanying cardiac defects.
MYOTONIC DYSTROPHY
Myotonic dystrophy type 1 (DM1, or Steinert’s disease) is one of the most common genetic disorders, affecting 1 in 8000 individuals. DM1 is associated with a trinucleotide expansion on chromosome 19. In subsequent generations within a family, increased expansion of this repeat sequence produces an earlier age of onset consistent with classic genetic anticipation, similar to neurodegenerative disorders such as Huntington’s or the spinocerebellar ataxias. The findings in DM1 are those of progressive muscle weakness and myotonia. Myotonia is characterized by a delay in muscle relaxation after normal contraction and by characteristic changes on electromyography. The typical electromyography manifestations are high-frequency muscle
fiber discharges of waxing and waning amplitude. Extramuscular findings include cardiac
atrioventricular heart block and cardiomyopathy, cataracts, testicular failure, disruption
of sleep, and profound fatigue (91). Neuropsychiatric abnormalities may also be present.
The trinucleotide expansion on chromosome 19 falls within the 3 end of the myotonic dystrophy protein kinase (DMPK) gene, and aspects of the cardiac dysfunction may relate to the disruption of the function of DM protein kinase. The trinucleotide expansion also encompasses the promoter of the adjacent SIX5 gene, and deletions of this gene recapitulate extramuscular aspects of the DM1-associated phenotype.
Myotonic dystrophy is associated with a nucleotide repeat expansion. The current favored hypothesis is that myotonia relates to a gain of function associated with the production and accumulation of CUG repeat–containing RNA within the nuclei of DM1 patients (Figure 9).
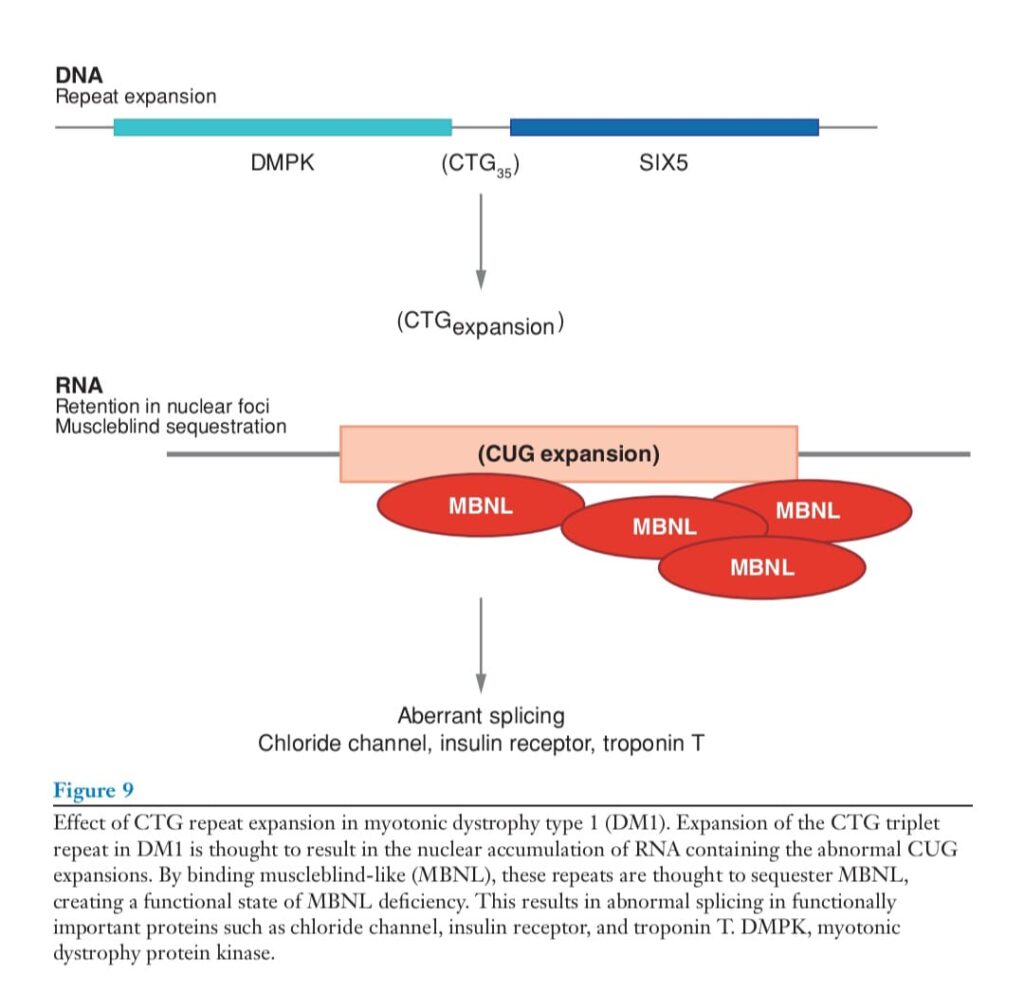
Transgenic overexpression of the trinucleotide repeat associated with an inconsequential gene (human skeletal actin) was sufficient to produce myotonia and muscle wasting (92). In this case, the transgene was expressed only in muscle, thereby limiting the pathology to muscle. This finding was extended further when it was observed that expanded, expressed repeats sequester the muscleblind proteins, limiting their normal participation in RNA binding (93, 94). The loss of muscleblind proteins results in abnormal splicing of a number of genes (95). Target
genes whose RNA is improperly spliced include the insulin receptor, the chloride channel, myotubularin, tau, and troponin T, proteins that play roles in the variable aspects of the phenotype of DM1. Mice with targeted gene disruption of muscleblind genes develop myotonia and have confirmed the role of muscleblind genes in specific deregulation of splicing (96). As it is this myotonia that defines the disorder, these animals display the typical, small, polyphasic
short-duration motor unit potentials as electromyography findings. Particularly interesting is that misregulation of splicing is not uniform, but rather, affects a subset of gene products.
A clear genotype-phenotype correlation does not exist, except in the broadest sense for myotonic dystrophy. For CTG nucleotide repeat expansions less than 400, there may be
some correlation with phenotype. For longer repeats, there is less correlation. Individuals
with profound expansions between two subsequent generations with maternal inheritance
are more likely to suffer congenital myotonic dystrophy. Congenital myotonic dystrophy is
a severe, neonatal-onset disorder that can be lethal or lead to very early onset of diseases
associated with cognitive impairment, in addition to profound muscle weakness. In general, CTG trinucleotide repeat expansions can be quite variable in their presentation with respect to the degree of muscle weakness. The pattern of muscle weakness in DM1 frequently involves the facial muscles, with ptosis, difficulty closing the eyes and mouth, and difficulty chewing being among the earlier signs. Weakness can affect any muscle and usually progresses. The histological phenotype of DM1 (Figure 7) on muscle biopsies is much more variable than that of DMD. Often there is very little evidence of myofiber necrosis. This is also reflected in often near-normal CK levels. Instead, the most prominent features are often internalized central nuclei and variation in fiber size, with atrophic changes that predominantly affect type I fibers and some compensatory type II fiber hypertrophy. Chronic changes in the form of fibrosis and fatty replacement develop at a much more variable rate than in DMD.
Myotonic Dystrophy Type 2
Not all patients with a DM-like phenotype showed an expansion in the chromosome 19
region, nor did these individuals link to this region genetically. A second locus on chromosome 3 was mapped and subsequently identified. DM2, like DM1, is associated with an expansion in untranslated RNA (97). The DM2 region arises from a CCTG tetranucleotide repeat expansion in zinc finger protein 9. The phenotype with DM2 repeat expansions may be milder, especially with respect to the cardiac phenotype, although cardiac involvement can occur (98, 99). Presently, evidence supports that DM2, like DM1, is associated with a similar RNA-mediated toxicity (100). Finally, there are clearly patients with a DM-like phenotype who do not have expansions at either the chromosome 19 or chromosome 3 locus, suggesting that other repeats can expand and be expressed highly enough to participate in similar processes.
FASCIOSCAPULOHUMERAL MUSCULAR DYSTROPHY
Fascioscapulohumeral muscular dystrophy (FSHD) is the third most common form of
inherited myopathy and is a dominantly inherited form of progressive muscle wasting
(101, 102). FSHD is striking for its predilection to involve discreet muscle groups. Muscles of the face are characteristically affected, with ptosis being a prominent feature. Scapular winging is common. Molecularly, FSHD is associated with a deletion that affects the subtelomeric region of chromosome 4q. A 3.3 kb repeated DNA sequence termed D4Z4 is normally found over a 50–300 kb region, with 11 to 150 copies of the repeat. In FSHD patients, there are typically fewer than 11 repeats present in this region, and this effective deletion is thought to have a positional
effect on neighboring genes. Most patients with an FSHD-like phenotype display a reduced number of D4Z4 repeats, and there appears to be little genetic heterogeneity when strict diagnostic criteria are used. Curiously, there is a similar repeat constellation at the
subtelomeric region of chromosome 10p, but in this case alteration of the repeats at this locus is not associated with a muscle phenotype or any other extraskeletal manifestation. The
prevailing theory is that the D4Z4 repeat produces the phenotype through a positional effect because D4Z4 contains a transcriptional silencer. A recent study directly examined this
hypothesis through overexpression of three potential target genes on 4q. FRG1 (FSHD
region gene 1) is located approximately 100 kb centromeric to the D4Z4 region. Overexpression using murine transgenesis of FRG1, but not FRG2 or ANT1, was sufficient to produce a myopathic phenotype consistent with the notion that overexpression of FRG1 is toxic to muscle and, therefore, pathogenic
in FSHD (103). These findings, although intriguing, may not fully account for the pathogenesis of FSHD because whether FRG1 is abnormally overexpressed in FSHD muscle remains in debate (104). In addition to these studies, the telomeric region of chromosome 4q is also found at
the nuclear periphery, unlike other telomeric regions (105). Although this position is not
thought to be different between FSHD mutant and normal cells, this nuclear positioning may further influence gene expression of neighboring genes and provide a distinct, potentially additional mechanism for positioneffect variegation
REGULATORS OF MUSCLE GROWTH: THERAPY BEYOND MUSCLE-WASTING DISORDERS
In addition to these disorders, there are other causes of muscle degeneration, and the most
common form of muscle wasting, sarcopenia, occurs with aging. The mechanisms that
underlie sarcopenia are probably multifold and likely include defects of muscle growth
or regeneration. Several distinct and important pathways that mediate growth and regeneration during development and in adult life, including mature or older animals, have been uncovered in recent years. There are at least two pathways being pursued as treatment strategies in muscle-wasting disorders. In both cases, the effects of these pathways are not specific to diseased muscle and may be expanded upon to treat sarcopenia. Myostatin, also known as growth and differentiation factor 8, is a member of the transforming growth factor β family of proteins
(106). Myostatin is a natural inhibitor of muscle growth and is highly expressed in muscle. Mice engineered to lack the myostatin gene display a profound increase in muscle
mass with more than a 200% increase in muscle size. Myostatin is processed to form a
dimer and binds the activin type IIB receptor and subsequently activates SMADs and affects gene expression. Myostatin can bind to follistatin in a latent complex. Mice overexpressing follistatin similarly show an increase in muscle mass (107). A number of naturally occurring alleles of myostatin loss of function have been described in large animals, leading to the double muscling phenotype in cattle. Myostatin shows promise as a therapeutic target because an antibody that inhibits muscle growth can lead to an increase in muscle mass in normal muscle (108). In addition, myostatin inhibition in the background of the mdx mouse that lacks dystrophin also produced larger and stronger muscles (109). Most recently, myostatin inhibition has also been achieved through peptide inhibition (110). In addition to its role in muscle hypertrophy, myostatin exerts an effect on the proliferation and differentiation of muscle precursor cells
or myoblasts.
Insulin-like Growth Factor-1
Insulin-like growth factor-1 (IGF-1) mediates growth of a number of somatic tissues, including muscle. Specific forms of IGF-1mediate muscle growth in the setting of normal and dystrophic muscle. Transgenic overexpression of IGF-1 leads to a profound increase in muscle mass, similar to what is seen with the deletion of the myostatin locus (111). Interestingly, this result is highly dependent on the specific splice form of IGF-1 used. IGF-1 may act locally as well. Viral delivery
of IGF-1 is also effective at generating enhanced muscle mass both normally and in the setting of dystrophic muscle (112). A small form of IGF-1, known as muscle growth factor, is thought to be very effective at increasing muscle mass when delivered locally (113).
SUMMARY
Genetic studies have identified a large number of distinct monogenic causes of muscular degeneration and muscular dystrophy. The phenotype associated with these disorders varies and includes the mildest weak ness to the most profound loss of skeletal muscle function. Subsets of genes implicated in the muscular dystrophies share specific pathologic defects. The DGC, including dystrophin and the sarcoglycans, is essential for the mechanosignaling maintenance of plasma membrane stability in muscle. Dysferlin and, potentially, caveolin facilitate membrane repair. Calpain, potentially titin, and TRIM32 may mediate protein turnover. Proteins of the Z band such as myotilin and telethonin may have diverse roles related to sarcomere stability and signaling. Nuclear membrane proteins mediate dystrophic pathology by
providing structural integrity to the nuclear membrane and potentially through epistatic
mechanisms. Myotonic dystrophy is associated with a toxic RNA pathology, whereas FSHD arises from a positional effect on gene expression. These diverse mechanisms lead to
the common pathway of muscle degeneration and weakness, and therapy aimed at stimulating muscle growth may be effective against these common pathways. Other approaches
to therapy, such as stop codon read-through, require the detailed specific knowledge of the
gene defect. Because skeletal muscle regeneration is ongoing and mediated by the fusion
of mononuclear stem cells, cell-based therapy may prove effective at introducing corrected
genes into the multinuclear syncytium that is mature muscle.
ACKNOWLEDGMENTS
EMM is supported by the Muscular Dystrophy Association, the Burroughs Wellcome Fund,
the Heart Research Foundation, and the National Institutes of Health.
LITERATURE CITED
- Mauro A. 1961. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 9:493–95
- Relaix F, Rocancourt D, Mansouri A, Buckingham M. 2005. A Pax3/Pax7-dependent
population of skeletal muscle progenitor cells. Nature 435:948–53 - LaBarge MA, Blau HM. 2002. Biological progression from adult bone marrow to
mononucleate muscle stem cell to multinucleate muscle fiber in response to injury. Cell
111:589–601 - Reviewed satellite cells as tissue-specific stem cells and provided new insights into the heterogeneous nature of this cell pool. 4. Morgan JE, Partridge TA. 2003. Muscle satellite cells. Int. J. Biochem. Cell Biol.
35:1151–56
IGF-1: insulin-like
growth factor-1
www.annualreviews.org • The Muscular Dystrophies 103
Annu. Rev. Pathol. Mech. Dis. 2007.2:87-109. Downloaded from www.annualreviews.org
by Drexel University on 01/13/13. For personal use only. - Kobayashi K, Nakahori Y, Miyake M, Matsumura K, Kondo-Iida E, et al. 1998. An
ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy.
Nature 394:388–92 - Yoshida A, Kobayashi K, Manya H, Taniguchi K, Kano H, et al. 2001. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase,
POMGnT1. Dev. Cell 1:717–24 - Beltran-Valero de Bernabe D, Currier S, Steinbrecher A, Celli J, van Beusekom E, et al.
- Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am. J. Hum. Genet. 71:1033–43
- Beltran-Valero de Bernabe D, Voit T, Longman C, Steinbrecher A, Straub V, et al. 2004.
Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg
syndrome. J. Med. Genet. 41:e61 - Mercuri E, Brockington M, Straub V, Quijano-Roy S, Yuva Y, et al. 2003. Phenotypic
spectrum associated with mutations in the fukutin-related protein gene. Ann. Neurol.
53:537–42 - Reviewed thenormal function of the dystroglycan complex and the alterations caused
by defective posttranslational processing. - Barresi R, Campbell KP. 2006. Dystroglycan: from biosynthesis to pathogenesis
of human disease. J. Cell Sci. 119:199–207 - Brancaccio A. 2005. Alpha-dystroglycan, the usual suspect? Neuromuscul. Disord. 15:825–
28 - Durbeej M, Henry MD, Campbell KP. 1998. Dystroglycan in development and disease.
Curr. Opin. Cell Biol. 10:594–601 - Gee SH, Blacher RW, Douville PJ, Provost PR, Yurchenco PD, Carbonetto S. 1993.
Laminin-binding protein 120 from brain is closely related to the dystrophin-associated
glycoprotein, dystroglycan, and binds with high affinity to the major heparin binding
domain of laminin. J. Biol. Chem. 268:14972–80 - Brockington M, Blake DJ, Prandini P, Brown SC, Torelli S, et al. 2001. Mutations in the
fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with
secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan.
Am. J. Hum. Genet. 69:1198–209 - Kondo-Iida E, Kobayashi K, Watanabe M, Sasaki J, Kumagai T, et al. 1999. Novel
mutations and genotype-phenotype relationships in 107 families with Fukuyama-type
congenital muscular dystrophy (FCMD). Hum. Mol. Genet. 8:2303–9 - Kirschner J, Bonnemann CG. 2004. The congenital and limb-girdle muscular dystrophies: sharpening the focus, blurring the boundaries. Arch. Neurol. 61:189–99
- Muntoni F. 2004. Journey into muscular dystrophies caused by abnormal glycosylation.
Acta Myol. 23:79–84 - Haliloglu G, Topaloglu H. 2004. Glycosylation defects in muscular dystrophies. Curr.
Opin. Neurol. 17:521–27 - Torelli S, Brown SC, Brockington M, Dolatshad NF, Jimenez C, et al. 2005. Sub-cellular
localisation of fukutin related protein in different cell lines and in the muscle of patients
with MDC1C and LGMD2I. Neuromuscul. Disord. 15:836–43 - Esapa CT, McIlhinney RA, Blake DJ. 2005. Fukutin-related protein mutations that cause
congenital muscular dystrophy result in ER-retention of the mutant protein in cultured
cells. Hum. Mol. Genet. 14:295–305 - Grewal PK, McLaughlan JM, Moore CJ, Browning CA, Hewitt JE. 2005. Characterization of the LARGE family of putative glycosyltransferases associated with dystroglycanopathies. Glycobiology 15:912–23
- Matsumoto H, Noguchi S, Sugie K, Ogawa M, Murayama K, et al. 2004. Subcellular
localization of fukutin and fukutin-related protein in muscle cells. J. Biochem. 135:709–12
104 McNally · Pytel
Annu. Rev. Pathol. Mech. Dis. 2007.2:87-109. Downloaded from www.annualreviews.org
by Drexel University on 01/13/13. For personal use only. - Guicheney P, Vignier N, Helbling-Leclerc A, Nissinen M, Zhang X, et al. 1997. Genetics
of laminin alpha 2 chain (or merosin) deficient congenital muscular dystrophy: from
identification of mutations to prenatal diagnosis. Neuromuscul. Disord. 7:180–86 - Pegoraro E, Marks H, Garcia CA, Crawford T, Mancias P, et al. 1998. Laminin alpha2
muscular dystrophy: genotype/phenotype studies of 22 patients. Neurology 51:101–10 - Miller RG, Hoffman EP. 1994. Molecular diagnosis and modern management of
Duchenne muscular dystrophy. Neurol. Clin. 12:699–725 - Chaturvedi LS, Mukherjee M, Srivastava S, Mittal RD, Mittal B. 2001. Point mutation
and polymorphism in Duchenne/Becker muscular dystrophy (D/BMD) patients. Exp.
Mol. Med. 33:251–56 - Sicinski P, Geng Y, Ryder-Cook AS, Barnard EA, Darlison MG, Barnard PJ. 1989.
The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science
244:1578–80 - Howard MT, Anderson CB, Fass U, Khatri S, Gesteland RF, et al. 2004. Readthrough of
dystrophin stop codon mutations induced by aminoglycosides. Ann. Neurol. 55:422–26 - Aminoglycoside antibiotics produce read-through expression of dystrophin by suppressing the effect of stop codon mutations.
- Barton-Davis ER, Cordier L, Shoturma DI, Leland SE, Sweeney HL. 1999. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice.
J. Clin. Invest. 104:375–81 - Hoogerwaard EM, Bakker E, Ippel PF, Oosterwijk JC, Majoor-Krakauer DF, et al. 1999.
Signs and symptoms of Duchenne muscular dystrophy and Becker muscular dystrophy
among carriers in The Netherlands: a cohort study. Lancet 353:2116–19 - Koenig M, Kunkel LM. 1990. Detailed analysis of the repeat domain of dystrophin reveals
four potential hinge segments that may confer flexibility. J. Biol. Chem. 265:4560–66 - Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW,
Campbell KP. 1992. Primary structure of dystrophin-associated glycoproteins linking
dystrophin to the extracellular matrix. Nature 355:696–702 - Dystrophin and the DGC form a strong mechanical coupling between the sarcolemma
and the sarcomeric actin. - Rybakova IN, Patel JR, Ervasti JM. 2000. The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J. Cell Biol.
150:1209–14 - Matsuda R, Nishikawa A, Tanaka H. 1995. Visualization of dystrophic muscle fibers in
mdx mouse by vital staining with Evans blue: evidence of apoptosis in dystrophin-deficient
muscle. J. Biochem. 118:959–64 - Straub V, Rafael JA, Chamberlain JS, Campbell KP. 1997. Animal models for muscular
dystrophy show different patterns of sarcolemmal disruption. J. Cell Biol. 139:375–85 - Gregorevic P, Blankinship MJ, Allen JM, Crawford RW, Meuse L, et al. 2004. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat.
Med. 10:828–34 - Alderton JM, Steinhardt RA. 2000. How calcium influx through calcium leak channels
is responsible for the elevated levels of calcium-dependent proteolysis in dystrophic myotubes. Trends Cardiovasc. Med. 10:268–72 - Ervasti JM, Ohlendieck K, Kahl SD, Gaver MG, Campbell KP. 1990. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 345:315–19
- Yoshida M, Ozawa E. 1990. Glycoprotein complex anchoring dystrophin to sarcolemma.
J. Biochem. 108:748–52 - Lapidos KA, Kakkar R, McNally EM. 2004. The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ. Res. 94:1023–31
- Lovering RM, Porter NC, Bloch RJ. 2005. The muscular dystrophies: from genes to
therapies. Phys. Ther. 85:1372–88
www.annualreviews.org • The Muscular Dystrophies 105
Annu. Rev. Pathol. Mech. Dis. 2007.2:87-109. Downloaded from www.annualreviews.org
by Drexel University on 01/13/13. For personal use only. - Loufrani L, Levy BI, Henrion D. 2002. Defect in microvascular adaptation to chronic
changes in blood flow in mice lacking the gene encoding for dystrophin. Circ. Res.
91:1183–89 - Sander M, Chavoshan B, Harris SA, Iannaccone ST, Stull JT, et al. 2000. Functional
muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children
with Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 97:13818–23 - Thomas GD, Shaul PW, Yuhanna IS, Froehner SC, Adams ME. 2003. Vasomodulation
by skeletal muscle-derived nitric oxide requires alpha-syntrophin-mediated sarcolemmal
localization of neuronal Nitric oxide synthase. Circ. Res. 92:554–60 - Ozawa E, Mizuno Y, Hagiwara Y, Sasaoka T, Yoshida M. 2005. Molecular and cell biology
of the sarcoglycan complex. Musc. Nerv. 32:563–76 - Zimprich A, Grabowski M, Asmus F, Naumann M, Berg D, et al. 2001. Mutations in
the gene encoding epsilon-sarcoglycan cause myoclonus-dystonia syndrome. Nat. Genet.
29:66–69 - Wheeler MT, Zarnegar S, McNally EM. 2002. Zeta-sarcoglycan, a novel component of
the sarcoglycan complex, is reduced in muscular dystrophy. Hum. Mol. Genet. 11:2147–54 - Bueno MR, Moreira ES, Vainzof M, Chamberlain J, Marie SK, et al. 1995. A common
missense mutation in the adhalin gene in three unrelated Brazilian families with a relatively mild form of autosomal recessive limb-girdle muscular dystrophy. Hum. Mol. Genet.
4:1163–67 - McNally EM, Passos-Bueno MR, Bonnemann CG, Vainzof M, de Sa Moreira E, et al.
- Mild and severe muscular dystrophy caused by a single gamma-sarcoglycan mutation. Am. J. Hum. Genet. 59:1040–47
- Heydemann A, Huber JM, Demonbreun A, Hadhazy M, McNally EM. 2005. Genetic
background influences muscular dystrophy. Neuromuscul. Disord. 15:601–9 - Vainzof M, Passos-Bueno MR, Canovas M, Moreira ES, Pavanello RC, et al. 1996. The
sarcoglycan complex in the six autosomal recessive limb-girdle muscular dystrophies.
Hum. Mol. Genet. 5:1963–69 - Hack AA, Lam MY, Cordier L, Shoturma DI, Ly CT, et al. 2000. Differential requirement
for individual sarcoglycans and dystrophin in the assembly and function of the dystrophinglycoprotein complex. J. Cell Sci. 113(Pt 14):2535–44 - Straub V, Ettinger AJ, Durbeej M, Venzke DP, Cutshall S, et al. 1999. ε-sarcoglycan
replaces α-sarcoglycan in smooth muscle to form a unique dystrophin-glycoprotein complex. J. Biol. Chem. 274:27989–96 - Coral-Vazquez R, Cohn RD, Moore SA, Hill JA, Weiss RM, et al. 1999. Disruption of
the sarcoglycan-sarcospan complex in vascular smooth muscle: a novel mechanism for
cardiomyopathy and muscular dystrophy. Cell 98:465–74 - Coronary artery vasospasm in sarcoglycandeficient animals is caused by extrinsic factors, not primary smooth muscle dysfunction.
- Wheeler MT, Allikian MJ, Heydemann A, Hadhazy M, Zarnegar S, McNally EM.
- Smooth muscle cell-extrinsic vascular spasm arises from cardiomyocyte degeneration in sarcoglycan-deficient cardiomyopathy. J. Clin. Invest. 113:668–75
- Anderson LV, Davison K, Moss JA, Young C, Cullen MJ, et al. 1999. Dysferlin is a plasma
membrane protein and is expressed early in human development. Hum. Mol. Genet. 8:855–
61 - Achanzar WE, Ward S. 1997. A nematode gene required for sperm vesicle fusion. J. Cell
Sci. 110:1073–81 - Ward S, Argon Y, Nelson GA. 1981. Sperm morphogenesis in wild-type and fertilizationdefective mutants of Caenorhabditis elegans. J. Cell Biol. 91:26–44
106 McNally · Pytel
Annu. Rev. Pathol. Mech. Dis. 2007.2:87-109. Downloaded from www.annualreviews.org
by Drexel University on 01/13/13. For personal use only. - Piccolo F, Moore SA, Ford GC, Campbell KP. 2000. Intracellular accumulation and
reduced sarcolemmal expression of dysferlin in limb-girdle muscular dystrophies. Ann.
Neurol. 48:902–12 - Selcen D, Stilling G, Engel AG. 2001. The earliest pathologic alterations in dysferlinopathy. Neurology 56:1472–81
- Dysferlin is a transmembrane protein important in calciumdependent repair of defects in the
sarcolemmal membrane. - Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, et al. 2003. Defective membrane
repair in dysferlin-deficient muscular dystrophy. Nature 423:168–72 - Cenacchi G, Fanin M, De Giorgi LB, Angelini C. 2005. Ultrastructural changes in dysferlinopathy support defective membrane repair mechanism. J. Clin. Pathol. 58:190–95
- Doherty KR, McNally EM. 2003. Repairing the tears: dysferlin in muscle membrane
repair. Trends Mol. Med. 9:327–30 - Fernandez-Chacon R, Konigstorfer A, Gerber SH, Garcia J, Matos MF, et al. 2001.
Synaptotagmin I functions as a calcium regulator of release probability. Nature 410:41–
49 - Davis DB, Doherty KR, Delmonte AJ, McNally EM. 2002. Calcium-sensitive phospholipid binding properties of normal and mutant ferlin C2 domains. J. Biol. Chem.
277:22883–88 - Davis DB, Delmonte AJ, Ly CT, McNally EM. 2000. Myoferlin, a candidate gene and
potential modifier of muscular dystrophy. Hum. Mol. Genet. 9:217–26 - Doherty KR, Cave A, Davis DB, Delmonte AJ, Posey A, et al. 2005. Normal myoblast
fusion requires myoferlin. Development 132:5565–75 - McNally EM, Ly CT, Rosenmann H, Mitrani Rosenbaum S, Jiang W, et al. 2000. Splicing
mutation in dysferlin produces limb-girdle muscular dystrophy with inflammation. Am.
J. Med. Genet. 91:305–12 - Gallardo E, Rojas-Garcia R, de Luna N, Pou A, Brown RHJ, Illa I. 2001. Inflammation
in dysferlin myopathy: immunohistochemical characterization of 13 patients. Neurology
57:2136–38 - Hoffman EP, Rao D, Pachman LM. 2002. Clarifying the boundaries between the inflammatory and dystrophic myopathies: insights from molecular diagnostics and microarrays.
Rheum. Dis. Clin. North Am. 28:743–57 - Weiler T, Bashir R, Anderson LV, Davison K, Moss JA, et al. 1999. Identical mutation
in patients with limb girdle muscular dystrophy type 2B or Miyoshi myopathy suggests a
role for modifier gene(s). Hum. Mol. Genet. 8:871–77 - Zatz M, Starling A. 2005. Calpains and disease. N. Engl. J. Med. 352:2413–23
- Guyon JR, Kudryashova E, Potts A, Dalkilic I, Brosius MA, et al. 2003. Calpain 3 cleaves
filamin C and regulates its ability to interact with gamma- and delta-sarcoglycans. Musc.
Nerv. 28:472–83 - Sorimachi H, Kinbara K, Kimura S, Takahashi M, Ishiura S, et al. 1995. Muscle-specific
calpain, p94, responsible for limb girdle muscular dystrophy type 2A, associates with
connectin through IS2, a p94-specific sequence. J. Biol. Chem. 270:31158–62 - Garvey SM, Rajan C, Lerner AP, Frankel WN, Cox GA. 2002. The muscular dystrophy
with myositis (mdm) mouse mutation disrupts a skeletal muscle-specific domain of titin.
Genomics 79:146–49 - Woodman SE, Sotgia F, Galbiati F, Minetti C, Lisanti MP. 2004. Caveolinopathies:
mutations in caveolin-3 cause four distinct autosomal dominant muscle diseases. Neurology
62:538–43 - Walter MC, Braun C, Vorgerd M, Poppe M, Thirion C, et al. 2003. Variable reduction
of caveolin-3 in patients with LGMD2B/MM. J. Neurol. 250:1431–38
www.annualreviews.org • The Muscular Dystrophies 107
Annu. Rev. Pathol. Mech. Dis. 2007.2:87-109. Downloaded from www.annualreviews.org
by Drexel University on 01/13/13. For personal use only. - Kudryashova E, Kudryashov D, Kramerova I, Spencer MJ. 2005. Trim32 is a ubiquitin
ligase mutated in limb girdle muscular dystrophy type 2H that binds to skeletal muscle
myosin and ubiquitinates actin. J. Mol. Biol. 354:413–24 - Hauser MA, Horrigan SK, Salmikangas P, Torian UM, Viles KD, et al. 2000. Myotilin
is mutated in limb girdle muscular dystrophy 1A. Hum. Mol. Genet. 9:2141–47 - Z-disc proteins may play an important role beyond the mechanics of the sarcomeres by
being involved in cell signaling. - Pyle WG, Solaro RJ. 2004. At the crossroads of myocardial signaling: the role of
Z-discs in intracellular signaling and cardiac function. Circ. Res. 94:296–305 - Wallgren-Pettersson C. 2002. Nemaline and myotubular myopathies. Semin. Pediatr.
Neurol. 9:132–44 - Segura-Totten M, Wilson KL. 2004. BAF: roles in chromatin, nuclear structure and
retrovirus integration. Trends Cell Biol. 14:261–66 - Ostlund C, Worman HJ. 2003. Nuclear envelope proteins and neuromuscular diseases.
Musc. Nerv. 27:393–406 - Helbling-Leclerc A, Bonne G, Schwartz K. 2002. Emery-Dreifuss muscular dystrophy.
Eur. J. Hum. Genet. 10:157–61 - Smith ED, Kudlow BA, Frock RL, Kennedy BK. 2005. A-type nuclear lamins, progerias
and other degenerative disorders. Mech. Ageing Dev. 126:447–60 - Defects in nuclear envelope proteins impair the mechanical function of the
nucleus and reduce the expression of mechanosensitive genes. - Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, et al. 2004. Lamin
A/C deficiency causes defective nuclear mechanics and mechanotransduction. J.
Clin. Invest. 113:370–78 - Lammerding J, Hsiao J, Schulze PC, Kozlov S, Stewart CL, Lee RT. 2005. Abnormal
nuclear shape and impaired mechanotransduction in emerin-deficient cells. J. Cell Biol.
170:781–91 - Bakay M, Wang Z, Melcon G, Schiltz L, Xuan J, et al. 2006. Nuclear envelope dystrophies
show a transcriptional fingerprint suggesting disruption of Rb-MyoD pathways in muscle
regeneration. Brain 129(4):996–1013 - Melcon G, Kozlov S, Cutler DA, Sullivan T, Hernandez L, et al. 2006. Loss of emerin at
the nuclear envelope disrupts the Rb1/E2F and MyoD pathways during muscle regeneration. Hum. Mol. Genet. 15:637–51 - Frock RL, Kudlow BA, Evans AM, Jameson SA, Hauschka SD, Kennedy BK. 2006.
Lamin A/C and emerin are critical for skeletal muscle satellite cell differentiation. Genes
Dev. 20:486–500 - Machuca-Tzili L, Brook D, Hilton-Jones D. 2005. Clinical and molecular aspects of the
myotonic dystrophies: a review. Musc. Nerv. 32:1–18 - Mankodi A, Logigian E, Callahan L, McClain C, White R, et al. 2000. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 289:1769–73
- Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, et al. 2000. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic
dystrophy. EMBO J. 19:4439–48 - Mankodi A, Urbinati CR, Yuan QP, Moxley RT, Sansone V, et al. 2001. Muscleblind
localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum.
Mol. Genet. 10:2165–70 - Ho TH, Charlet BN, Poulos MG, Singh G, Swanson MS, Cooper TA. 2004. Muscleblind
proteins regulate alternative splicing. EMBO J. 23:3103–12 - Muscleblindlike deleted mice show the same splicing abnormalities found in DM patients.
Sequestration of muscleblind-like through RNA binding can cause DM. - Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, et al. 2003. A
muscleblind knockout model for myotonic dystrophy. Science 302:1978–80 - Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, et al. 2001. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293:864–67
108 McNally · Pytel
Annu. Rev. Pathol. Mech. Dis. 2007.2:87-109. Downloaded from www.annualreviews.org
by Drexel University on 01/13/13. For personal use only. - Ranum LP, Day JW. 2002. Myotonic dystrophy: clinical and molecular parallels between
myotonic dystrophy type 1 and type 2. Curr. Neurol. Neurosci. Rep. 2:465–70 - Schoser BG, Ricker K, Schneider-Gold C, Hengstenberg C, Durre J, et al. 2004. Sudden
cardiac death in myotonic dystrophy type 2. Neurology 63:2402–4 - Day JW, Ricker K, Jacobsen JF, Rasmussen LJ, Dick KA, et al. 2003. Myotonic dystrophy
type 2: molecular, diagnostic and clinical spectrum. Neurology 60:657–64 - Tupler R, Gabellini D. 2004. Molecular basis of facioscapulohumeral muscular dystrophy.
Cell Mol. Life Sci. 61:557–66 - Tawil R. 2004. Facioscapulohumeral muscular dystrophy. Curr. Neurol. Neurosci. Rep.
4:51–54 - Deletions may cause FSHD by inappropriate overexpression of FRG1 through positional changes on gene regulation.
- Gabellini D, D’Antona G, Moggio M, Prelle A, Zecca C, et al. 2006. Facioscapulohumeral muscular dystrophy in mice overexpressing FRG1. Nature 439:973–77
- van Deutekom JC, Lemmers RJ, Grewal PK, van Geel M, Romberg S, et al. 1996.
Identification of the first gene (FRG1) from the FSHD region on human chromosome
4q35. Hum. Mol. Genet. 5:581–90 - Masny PS, Bengtsson U, Chung SA, Martin JH, van Engelen B, et al. 2004. Localization
of 4q35.2 to the nuclear periphery: Is FSHD a nuclear envelope disease? Hum. Mol. Genet.
13:1857–71 - Lee SJ, McPherron AC. 1999. Myostatin and the control of skeletal muscle mass. Curr.
Opin. Genet. Dev. 9:604–7 - Lee SJ, McPherron AC. 2001. Regulation of myostatin activity and muscle growth. Proc.
Natl. Acad. Sci. USA 98:9306–11 - Whittemore LA, Song K, Li X, Aghajanian J, Davies M, et al. 2003. Inhibition of myostatin in adult mice increases skeletal muscle mass and strength. Biochem. Biophys. Res.
Commun. 300:965–71 - Bogdanovich S, Krag TO, Barton ER, Morris LD, Whittemore LA, et al. 2002. Functional improvement of dystrophic muscle by myostatin blockade. Nature 420:418–21
- Bogdanovich S, Perkins KJ, Krag TO, Whittemore LA, Khurana TS. 2005. Myostatin
propeptide-mediated amelioration of dystrophic pathophysiology. FASEB J. 19:543–49 - Musaro A, McCullagh KJ, Naya FJ, Olson EN, Rosenthal N. 1999. IGF-1 induces skeletal
myocyte hypertrophy through calcineurin in association with GATA-2 and NF-ATc1.
Nature 400:581–85 - Barton ER, Morris L, Musaro A, Rosenthal N, Sweeney HL. 2002. Muscle-specific
expression of insulin-like growth factor I counters muscle decline in mdx mice. J. Cell
Biol. 157:137–48 - Yang SY, Goldspink G. 2002. Different roles of the IGF-I Ec peptide (MGF) and mature
IGF-I in myoblast proliferation and differentiation. FEBS Lett. 522:156–60
www.annu
SIDE NOTES
Annu. Rev. Pathol. Mech. Dis. 2007.
2:87–109
The Annual Review of Pathology: Mechanisms of Disease is online at pathmechdis.annualreviews.org
This article’s doi: 10.1146/annurev.pathol.2.010506.091936 Copyright c 2007 by Annual Reviews. All rights reserved 1553-4006/07/0228-0087$20.0
Muscular dystrophy:
genetically linked muscle disease characterized by progressive destruction of muscle tissue
Sarcomere: basic functional unit of muscle contraction consisting of actin, myosin, and
associated proteins
Sarcolemma: the specialized plasma membrane of the individual myofibers in skeletal muscle
Satellite cell: pool of localized tissue stem cells in the skeletal muscle important for its development and regenerative potential CMD: congenital muscular dystrophy
nNOS: neuronal nitric oxide synthase
LGMD: limb-girdle muscular dystrophy
EDMD: Emery-Dreifuss muscular dystrophy Nuclear membrane proteins: proteins that may play a role in mechanical functions and gene regulation
Myotonia: delayed relaxation after voluntary muscle contraction associated with characteristic
electrophysiological changes Nucleotide repeat expansion: expansion of normal repeats that alters gene expression or causes abnormal RNA or protein accumulation
FSHD: fascioscapulohumeral muscular dystrophy
Sarcopenia: age-related loss of muscle mass that may interfere with daily living
IGF-1: insulin-like growth factor-1